



Response to Lipoprotein Particle Pathway: Key Genes, Lipid Metabolism, and CRISPR KO Cell Models for Cardiovascular Disease Research

01
Overview of the Lipoprotein Response Pathway
● What is the Lipoprotein Response?
The Response to Lipoprotein Particle Pathway refers to the key biological processes by which cells or organisms react to stimulation by lipoprotein particles such as LDL, HDL, and VLDL (Gene Ontology term: GO:0055094). Lipoproteins are responsible for transporting lipids, including cholesterol, triglycerides, and phospholipids, through the bloodstream.
Maintaining their metabolic homeostasis is essential for cellular function, energy balance, and endocrine regulation. Disruption of lipoprotein metabolism can directly lead to major metabolic diseases, including atherosclerosis, hyperlipidemia, non-alcoholic fatty liver disease (NAFLD), obesity, and type 2 diabetes.
● Regulatory Network of the Lipoprotein Response
The Response to Lipoprotein Particle Pathway regulates multiple biological processes, including lipid uptake, inflammatory responses, cholesterol homeostasis, and innate immune signaling. When lipoprotein particles bind to cell surface receptors, such as LDLR, CD36, and TLR4, they activate a series of downstream signaling cascades involving molecules like MYD88, AKT1, PPARG, and SREBF2.
This leads to changes in gene expression, metabolic reprogramming, and cellular functions such as foam cell formation and cholesterol efflux. In functional genomics analyses, related GO terms show high statistical significance (FDR = 1.11 × 10⁻⁵), confirming the central role of this pathway in lipid metabolism regulation.
02
Molecular Mechanisms of the Lipoprotein Response: From Cholesterol Metabolism to Inflammatory Signaling
● Balance and Imbalance in Cholesterol Metabolism
Cellular cholesterol homeostasis is maintained through the coordinated regulation of uptake, synthesis, and efflux. Under physiological conditions, LDL is cleared via LDLR-mediated endocytosis, while the SREBP pathway provides negative feedback to control cholesterol synthesis.
In pathological states, oxidized LDL (ox-LDL) is taken up in large quantities by macrophages through scavenger receptors such as CD36. When this exceeds the cholesterol efflux capacity mediated by ABCA1, cholesterol esters accumulate, leading to foam cell formation.
Excess cholesterol transported to the endoplasmic reticulum triggers ER stress, which activates the NLRP3 inflammasome and promotes the release of IL-1β and IL-18—creating a vicious cycle between lipid dysregulation and chronic inflammation.
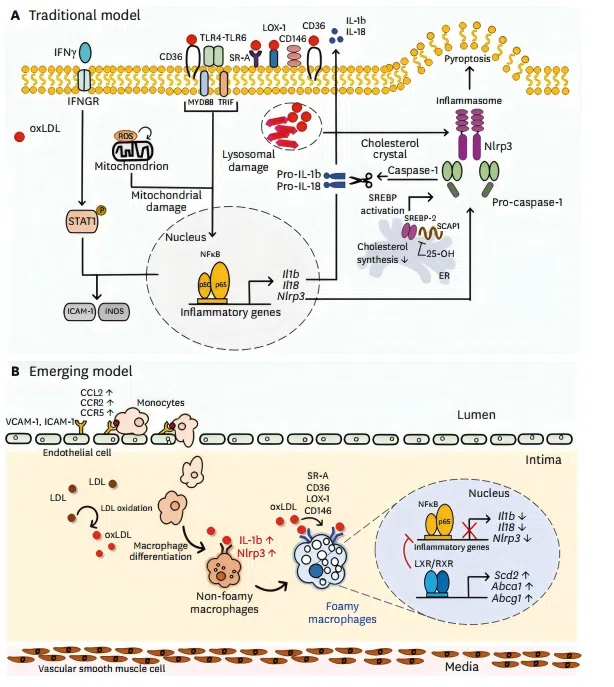 Figure 1. Phenotypic changes of macrophages by lipid uptake in atherosclerotic lesion.
Figure 1. Phenotypic changes of macrophages by lipid uptake in atherosclerotic lesion. ● Chronic Inflammation in the Arterial Wall
Oxidized phospholipids (OxPLs) carried by lipoproteins act as damage-associated molecular patterns (DAMPs). They are recognized by pattern recognition receptors such as TLR4, activating the MyD88-dependent NF-κB pathway.
This upregulates adhesion molecules including VCAM-1 and ICAM-1, promoting monocyte recruitment and differentiation. Once macrophages engulf lipoproteins, they further amplify inflammatory signals, accelerating plaque formation and destabilization.
● Signal Transduction and Metabolic Integration
Lipoprotein metabolism engages in extensive crosstalk with insulin signaling. Intracellular cholesterol levels regulate metabolic gene expression through nuclear receptors such as LXR and PPAR.
Lipid overload inhibits insulin signaling via diacylglycerol (DAG) and ceramides, resulting in peripheral insulin resistance. Dysregulation of the PI3K/Akt and AMPK pathways further exacerbates lipid accumulation, driving the development of obesity, type 2 diabetes, and non-alcoholic fatty liver disease.
03
Lipoprotein Response Dysregulation and Disease
● Atherosclerosis and Cardiovascular Disease
The deposition of low-density lipoprotein (LDL) in the arterial wall is the initiating event of atherosclerotic plaque formation. It triggers vascular dysfunction and immune dysregulation, promoting plaque progression.
Triglyceride-rich lipoproteins (TRLs) and their remnant particles exhibit even stronger atherogenic potential than LDL. Studies have shown that CD36 and TLR4 synergistically activate the NLRP3 inflammasome to promote foam cell formation, while UCHL1 stabilizes CD36 through deubiquitination, further aggravating atherosclerotic pathology.
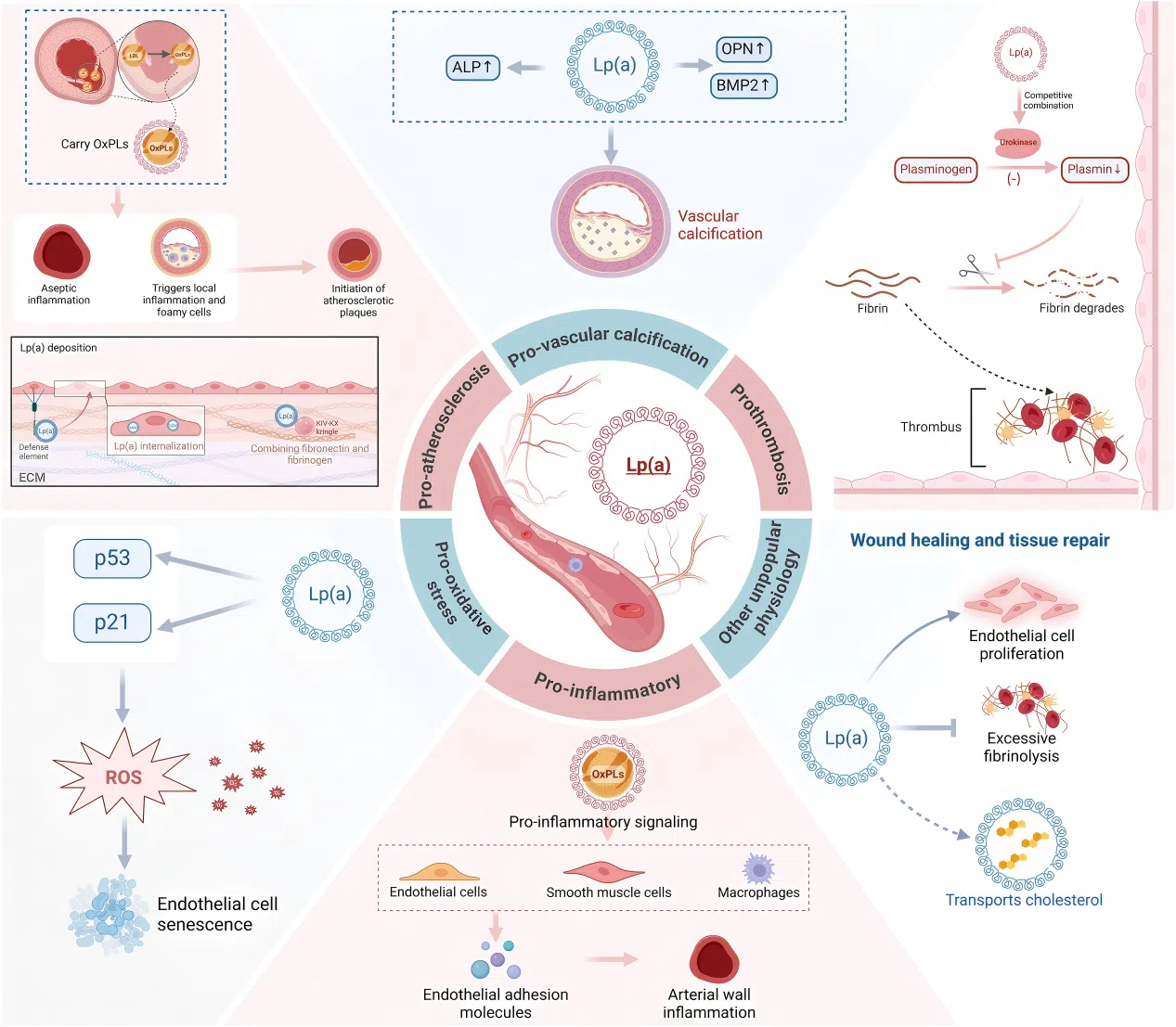 Figure 2. The pathophysiological mechanisms of Lp(a)
Figure 2. The pathophysiological mechanisms of Lp(a) 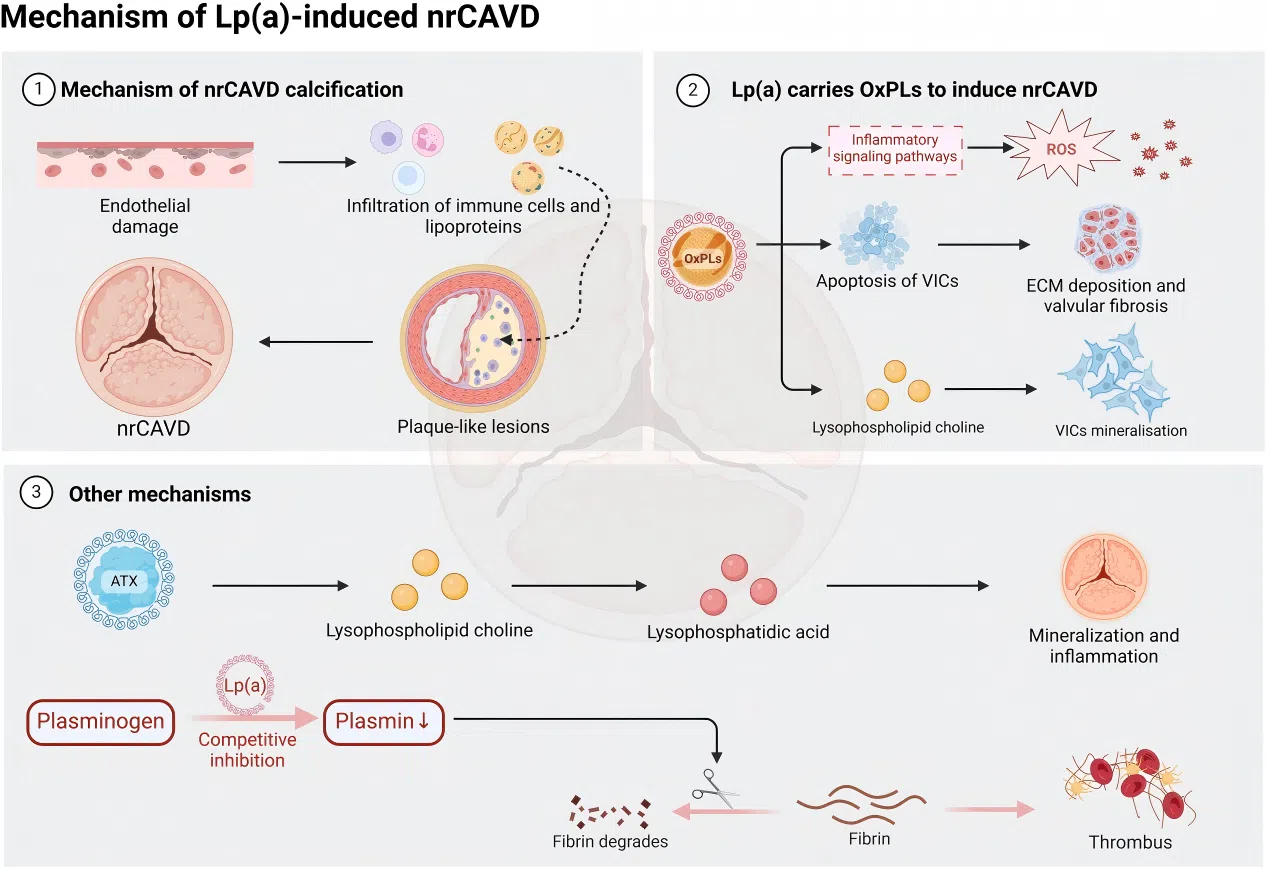 Figure 3. The impact of Lp(a) on nrCAVD pathogenesis
Figure 3. The impact of Lp(a) on nrCAVD pathogenesis ● Metabolic Syndrome and Neurodegenerative Diseases
Dysregulation of the lipoprotein response is also closely linked to hypercholesterolemia, non-alcoholic fatty liver disease, type 2 diabetes, and Alzheimer's disease.
For example, abnormal palmitoylation of CD36 can cause severe lipid overload in hepatocytes; mutating the palmitoylation sites can reduce hepatic triglyceride deposition by up to 50%.
These disease associations highlight the significant therapeutic potential of targeting the lipoprotein response pathway.
04
Research Strategies and Therapeutic Applications
● Current Treatment Strategies
Current intervention strategies targeting lipoprotein metabolism mainly include:
1. LDL-C reduction: Statins and PCSK9 inhibitors;
2. Anti-inflammatory therapies: Inhibitors targeting the TLR4/CD36 pathway;
3. Promotion of cholesterol efflux: ABCA1 agonists and HDL-mimetic peptides;
4. Novel LXR inverse agonists: TLC-2716 reduced postprandial remnant cholesterol by 61% in Phase I clinical trials, opening a promising new direction for the treatment of dyslipidemia.
● Value of CRISPR KO Cell Models
Gene knockout cell models allow researchers to precisely dissect the molecular mechanisms of the lipoprotein response in a clean genetic background, providing strong support for drug target validation and the construction of in vitro disease models.
CRISPR gene editing technology has been successfully used to generate knockout cell lines for multiple key genes, including LDLR, CD36, TLR4, ABCA1, PPARG, SREBF2, MYD88, TREM2, AKT1, and NPC1. These models serve as powerful tools for atherosclerosis research, metabolic inflammation studies, and drug screening.
Utilizing gene knockout cell models enables in-depth investigation into the essential roles of specific genes in the Response to Lipoprotein Particle Pathway, thereby accelerating new drug development and translational research.
EDITGENE has launched off-the-shelf KO cell lines covering over 40 core genes to support your metabolic disease research.
FAQ
Q1: I want to study the function of LDLR in specific primary macrophages, but this cell type is not listed.
We offer custom services. You can provide your target cell line (e.g., human primary macrophages, mouse bone marrow-derived macrophages, etc.), and we will handle the entire process from gene editing to monoclonal validation.
Q2: Can these KO cells be used for in vivo atherosclerosis model studies?
Yes. Many customers have used our LDLR KO, CD36 KO, or TLR4 KO macrophages or hepatocytes by inoculating them into mice to investigate the effects of gene deletion on lipid uptake, foam cell formation, plaque stability, and inflammatory infiltration.
Q3: I plan to perform high-throughput drug screening and need large quantities of cells from the same batch. Can you meet this requirement?
Yes. All our off-the-shelf cells are supported by master cell banks and working cell banks. We can supply large volumes of batch-consistent cells to ensure uniformity and comparability of your screening data.
References
[1]. Moore, K. J., & Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell, 145(3), 341-355.
[2]. TLC-2716 phase 1 trial for dyslipidemia. Nature Medicine, 2026;32:883-893.
[3]. Libby, P., et al. (2021). Atherosclerosis. Nature Reviews Disease Primers, 5(1), 56.
[4]. Soppert, J., et al. (2020). Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Advanced Drug Delivery Reviews, 159, 4-33.
Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com
Tag
Share







