















-
CRISPR Knockout KitCRISPR Point Mutation KitKI Enhancer Drug
-
Precision Mutation Cell PanelsKnock-in cell lineWild Type Cell Line
-
-
 Cardiovascular - Nitric Oxide Metabolic Pathway
Cardiovascular - Nitric Oxide Metabolic Pathway
Nitric oxide (NO) is a key signaling molecule in the cardiovascular, nervous, and immune systems. Its bioavailability depends not only on synthesis rate but also on the metabolic balance between clearance and conversion. Dysregulated NO metabolism is closely associated with pulmonary hypertension, ischemia-reperfusion injury, atherosclerosis, and neurodegenerative diseases. Gene knockout cell models targeting key regulators of NO metabolism enable precise investigation of NO clearance mechanisms and provide reliable tools for therapeutic target validation and oxidative stress-related drug discovery.
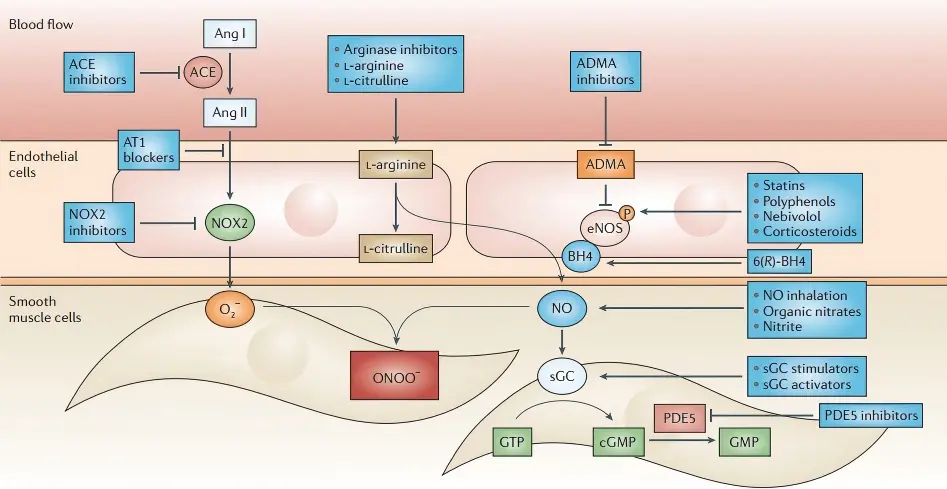
Nitric oxide (NO) metabolic clearance is mainly regulated through three pathways controlling its bioavailability:
① reaction with superoxide (O₂⁻) to generate peroxynitrite (ONOO⁻), leading to protein nitration damage;
② binding of NO with oxyhemoglobin to produce nitrate (NO₃⁻) and methemoglobin;
③ degradation of S-nitrosothiols (SNO) by GSNOR.
Key regulatory factors include: SOD1/2, which eliminate O₂⁻ to protect NO bioavailability; GCH1, which synthesizes BH4 to maintain eNOS coupling; arginases (Arg1/2), which compete for the substrate L-arginine; GSNOR, which degrades SNO; and hemoglobin (HBB), which scavenges NO.
When dysregulated, SOD deficiency accelerates NO depletion, GCH1 insufficiency causes eNOS uncoupling (producing O₂⁻ instead of NO), Arg1/2 overexpression suppresses NO synthesis, and GSNOR imbalance disrupts nitrosylation homeostasis.
Corresponding gene knockout cell models provide clean genetic backgrounds for dissecting NO metabolic mechanisms and screening compounds that regulate NO half-life and redox balance, offering valuable tools for cardiovascular, neurological, and metabolic disease research.
(Lundberg et al., Cell Metab, 2015)
The biological availability of nitric oxide (NO) depends not only on its synthesis but also on its metabolic fate through clearance and conversion pathways. Dysregulation of key metabolic regulators—including SOD, GCH1, arginases, GSNOR, and hemoglobin—can accelerate NO depletion or generate toxic metabolites, contributing to diseases such as pulmonary hypertension, ischemia-reperfusion injury, atherosclerosis, diabetic endothelial dysfunction, hemolysis-associated vasculopathy, asthma, cancer, and neurodegenerative disorders.
These disease associations highlight the therapeutic potential of targeting the NO metabolic pathway. Gene knockout cell models provide powerful tools for studying the regulatory functions of NO metabolic factors under pathological conditions and support mechanism studies, target validation, and drug discovery.
· Oxidative Stress and Cardiovascular Disease Models
Used to investigate the effects of SOD1/SOD2 knockout on superoxide accumulation and NO bioavailability, as well as to evaluate antioxidants or SOD mimetics.
· eNOS Coupling and Endothelial Dysfunction Models
Used to study BH4 deficiency and eNOS uncoupling caused by GCH1 knockout and to screen small molecules that restore eNOS function.
· Substrate Competition and Metabolic Disease Models
Used to investigate the effects of Arg1/Arg2 knockout on L-arginine metabolic flux and explore NO regulation in wound healing, tumor immunity, and metabolic syndrome.
· Nitrosylation Homeostasis and Signal Transduction Models
Used to study the impact of GSNOR knockout on the S-nitrosylated proteome and cellular signaling, providing screening platforms for GSNOR-targeting therapeutics in asthma, cancer, and related diseases.
EDITGENE’s NO metabolic pathway knockout cell line library focuses on key regulators involved in nitric oxide clearance and conversion. We provide validated gene knockout cell models for investigating oxidative stress, eNOS uncoupling, substrate competition, and nitrosylation imbalance-related disease mechanisms. Both ready-to-use and customized knockout cell services are available to support diverse research needs in redox biology and metabolism.
-
Cat.No: EDC90437
Species: Human
Cell Name: HEK293
Gene Name: DRD2
Gene ID: 1813
Specs: 1×10⁶cells
-
Cat.No: EDC10177
Species: Human
Cell Name: HeLa
Gene Name: GTPBP2
Gene ID: 54676
Specs: 1×10⁶cells
-
Cat.No: EDC09412
Species: Human
Cell Name: HAP1
Gene Name: TESK1
Gene ID: 7016
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ2237
Species: Human
Cell Name: HEK293
Gene Name: DRD4
Gene ID: 1815
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ2726
Species: Human
Cell Name: HEK293
Gene Name: DRD3
Gene ID: 1814
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ3388
Species: Human
Cell Name: HEK293
Gene Name: CSDE1
Gene ID: 7812
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ5916
Species: Human
Cell Name: HEK293
Gene Name: TESK1
Gene ID: 7016
Specs: 1×10⁶cells
-
Cat.No: EDC09659
Species: Human
Cell Name: HEK293
Gene Name: GTPBP2
Gene ID: 54676
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ12146
Species: Human
Cell Name: HEK293
Gene Name: CNOT6
Gene ID: 57472
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ25079
Species: Human
Cell Name: A-549
Gene Name: CSDE1
Gene ID: 7812
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ25080
Species: Human
Cell Name: HeLa
Gene Name: CSDE1
Gene ID: 7812
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ23690
Species: Human
Cell Name: HCT 116
Gene Name: CSDE1
Gene ID: 7812
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ29445
Species: Human
Cell Name: A-549
Gene Name: TESK1
Gene ID: 7016
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ29446
Species: Human
Cell Name: HCT 116
Gene Name: TESK1
Gene ID: 7016
Specs: 1×10⁶cells
-
Cat.No: EDJ-KQ29447
Species: Human
Cell Name: HeLa
Gene Name: TESK1
Gene ID: 7016
Specs: 1×10⁶cells
- 1
- 2
- Next Page »
Subscribe
You can unsubscribe from these communications at any time. For more information on how to unsubscribe, our privacy practices, and how we are committed to protecting and respecting your privacy, please review our Privacy Policy.
By clicking submit below, you consent to allow EDITGENE to store and process the personal information submitted above to provide you the content requested.
