















How Lipoprotein Particles Shape Lipid Metabolism and Disease: From Cellular Response Mechanisms to CRISPR KO Models

When the metabolic balance of lipoprotein particles in the blood is disrupted, it triggers chronic inflammation—the common root of atherosclerosis, metabolic syndrome, and even neurodegenerative diseases.
Circulating lipoprotein particles are the primary carriers of cholesterol and triglycerides: low-density lipoprotein (LDL) delivers cholesterol to tissues, while high-density lipoprotein (HDL) recycles it via reverse transport. Together, they maintain the delicate balance of lipid metabolism.
When this balance is broken—such as through oxidative deposition of LDL or dysfunction of HDL—damage-associated molecular patterns (DAMPs) like oxidized phospholipids (OxPLs) activate innate immune signaling, driving foam cell formation and metabolic inflammation. Pathway enrichment analysis shows that lipoprotein response pathways are significantly enriched in functional genomics (FDR = 1.11×10⁻⁵), further confirming their role as key regulatory hubs.
With CRISPR gene editing technology, it is now possible to precisely dissect the key regulators of lipoprotein metabolism, offering new avenues for targeted intervention in related diseases.
Understanding how imbalances in the lipoprotein system lead to disease requires a stepwise analysis from four levels: particle structure, metabolic pathways, immune activation, and gene editing validation.
01
Structural Dynamics and Biological Functions of Lipoprotein Particles
The metabolic homeostasis of lipoproteins is essential for maintaining cellular function, energy balance, and endocrine regulation.
Apolipoproteins not only help stabilize particle structure but also act as ligands that interact with receptors such as LDLR, SR-BI, and CD36, thereby regulating tissue-specific lipid uptake and clearance. ApoB-containing lipoproteins—including LDL, VLDL, and lipoprotein(a)—play a central role in the initiation and progression of atherosclerosis.
Lipoprotein metabolism involves three major dynamic pathways:
· Exogenous pathway: transport of dietary lipids via chylomicrons;
· Endogenous pathway: hepatic synthesis of VLDL, followed by hydrolysis by lipoprotein lipase to generate IDL and LDL;
· Reverse cholesterol transport pathway: HDL-mediated return of cholesterol from peripheral tissues to the liver.
It is worth emphasizing that cardiovascular protection depends on the subclass composition and functional status of HDL particles, not simply on plasma HDL‑C levels. Even with normal particle size, dysfunctional HDL loses its anti‑atherogenic activity.
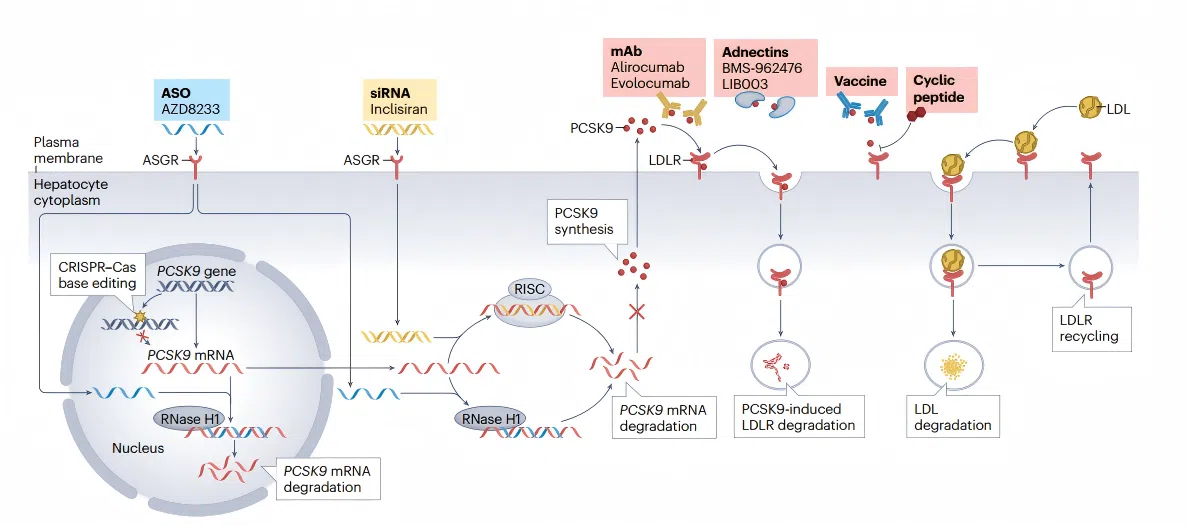 Figure 1. Overview of lipid metabolism pathways and emerging therapeutic targets for cardiovascular disease
Figure 1. Overview of lipid metabolism pathways and emerging therapeutic targets for cardiovascular disease 02
oxLDL and Foam Cell Formation: The Central Role of CD36
A core pathological feature of atherosclerotic plaques is the accumulation of lipid‑laden foam cells. These are derived primarily from macrophages that have taken up large amounts of oxidized low‑density lipoprotein (oxLDL), though vascular smooth muscle cells (VSMCs) can also transform into foam cells.
Macrophage uptake of oxLDL is a critical step in plaque initiation, and the scavenger receptor CD36 plays a dominant role in this process. CD36 recognizes oxidized phospholipids (OxPLs) on the surface of oxLDL, mediating massive oxLDL internalization and leading to intracellular cholesteryl ester accumulation.
Moreover, oxLDL induces pyruvate kinase M2 (PKM2)‑dependent mitochondrial reactive oxygen species production, which enhances macrophage phagocytic activity and further amplifies inflammatory responses. CD36 deficiency significantly reduces oxLDL‑induced phagocytic responses and suppresses foam cell formation.
While foam cell formation provides the "building material" for arterial plaques, persistent inflammatory signaling is the direct driver of plaque instability. The next section will explore lipoprotein‑induced inflammatory pathways.
03
Cholesterol Efflux and Reverse Cholesterol Transport
ATP-binding cassette transporter A1 (ABCA1) is the key rate‑limiting factor regulating cellular cholesterol efflux. By binding to lipid‑poor apolipoprotein A‑I (apoA‑I) and promoting its phospholipidation, ABCA1 initiates the formation of nascent HDL particles—the first step in reverse cholesterol transport (RCT). Reduced ABCA1 activity directly leads to foam cell accumulation.
Under cholesterol‑overloaded conditions, ABCA1 transports cholesterol to lipid‑poor apoA‑I, while ABCG1 mediates secondary transfer to mature HDL particles. Together, these two transporters work synergistically to maintain cholesterol homeostasis.
Of note, HDL often becomes dysfunctional in diabetes, exhibiting reduced cholesterol efflux capacity and diminished anti‑inflammatory activity. This dysfunction represents a key link connecting disturbed lipoprotein metabolism with insulin resistance.
04
The LDLR-PCSK9 Axis: A Key Regulatory Node in Cholesterol Metabolism
The LDL receptor (LDLR) is the core molecule that maintains plasma cholesterol homeostasis, primarily by clearing LDL cholesterol from the liver.
PCSK9 acts as a negative regulator of LDLR: upon binding to LDLR, it promotes lysosomal degradation of the receptor, thereby reducing LDLR surface expression and recycling.
Genetic variants in PCSK9 have a marked impact on plasma LDL‑C levels, and PCSK9 inhibitors have become an important therapeutic strategy for cardiovascular disease.
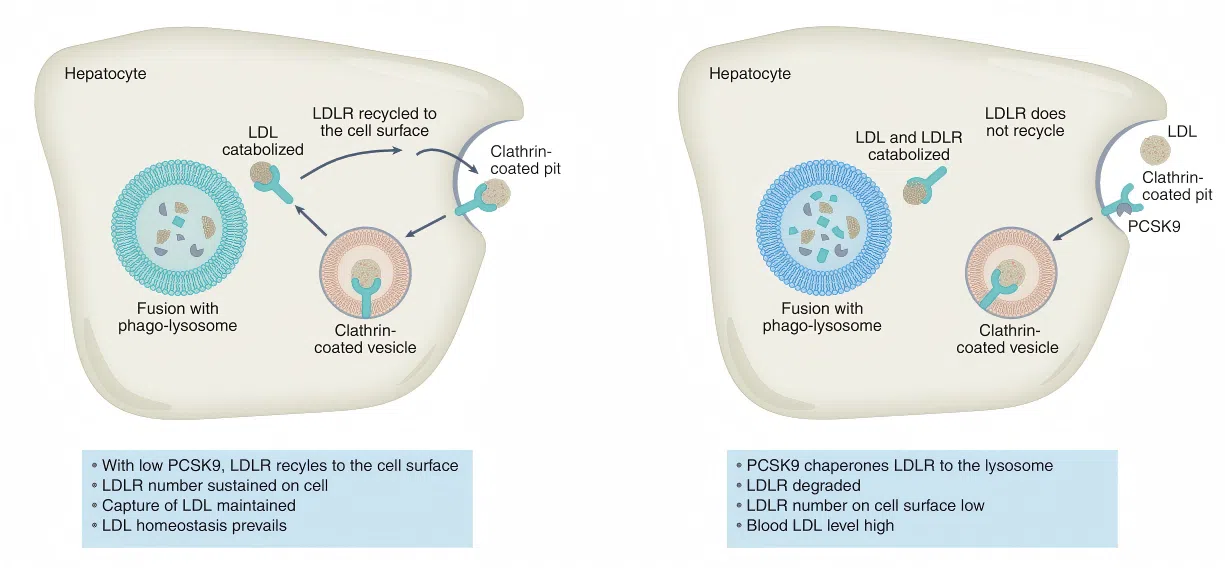 Figure 2. The PCSK9-LDLR axis: from LDL receptor degradation to cardiovascular therapy
Figure 2. The PCSK9-LDLR axis: from LDL receptor degradation to cardiovascular therapy Lipoprotein Metabolic Dysregulation and Diabetes
Lipoprotein metabolic dysregulation is deeply involved in the development and progression of type 2 diabetes.
Under insulin‑resistant conditions, adipose tissue lipolysis increases, leading to elevated free fatty acid release, which promotes hepatic VLDL overproduction and results in an atherogenic lipid profile (hypertriglyceridemia, low HDL‑C, and increased small, dense LDL). At the same time, chronic hyperglycemia accelerates LDL oxidation through enhanced oxidative stress and glycation, promoting oxLDL formation and foam cell generation.
Intervention strategies targeting lipoprotein metabolism—such as PCSK9 inhibitors and SGLT2 inhibitors—have demonstrated significant benefits in improving cardiovascular outcomes in patients with diabetes.
Lipoprotein‑Induced Inflammatory Signaling Pathways
Oxidized phospholipids (OxPLs) carried by oxLDL act as endogenous damage‑associated molecular patterns (DAMPs). They are recognized by Toll‑like receptor 4 (TLR4), which activates the MyD88‑dependent NF‑κB pathway, upregulating adhesion molecules such as VCAM‑1 and ICAM‑1, thereby promoting monocyte recruitment and differentiation.
TLR4 signaling and its adaptor molecule MyD88 directly participate in the pathogenesis of atherosclerosis. Infiltrating macrophages further amplify inflammatory signals, accelerating plaque formation and instability. Statins can shift the immune response toward an anti‑inflammatory phenotype by inhibiting the TLR4/MyD88/NF‑κB signaling pathway.
Lipoprotein Metabolic Dysregulation and Neurodegenerative Disease
Abnormal lipoprotein metabolism is also associated with Alzheimer’s disease. The apolipoprotein E (APOE) ε4 allele is a strong risk factor for sporadic Alzheimer’s disease, contributing to disease progression by affecting cerebral cholesterol transport and β‑amyloid clearance.
Triggering receptor expressed on myeloid cells 2 (TREM2) in microglia links peripheral lipoprotein metabolic disturbances to central neuroinflammation by regulating lipid sensing and metabolism.
05
Application of CRISPR Knockout Cell Models in Lipoprotein Research
For each of the key molecules involved in the pathogenic pathways described above—including CD36, ABCA1, LDLR, TLR4, MYD88, APOE, and others—CRISPR knockout (KO) cell models can be used to perform loss‑of‑function validation, thereby clarifying their essential roles in lipoprotein responses.
Gene‑edited KO cell models allow precise dissection of molecular mechanisms in a clean genetic background, supporting drug target validation and the construction of in vitro disease models. Common application scenarios include:
· Atherosclerosis and cardiovascular disease models: Using LDLR KO, CD36 KO, or TLR4 KO macrophage or hepatocyte cell lines to investigate the necessity of lipoprotein metabolism genes in oxLDL uptake, foam cell formation, and cholesterol efflux.
· Metabolic inflammation and insulin resistance: MYD88 KO or TLR4 KO cells can be used to dissect how lipoproteins influence insulin sensitivity via innate immune signaling pathways.
· Cholesterol homeostasis regulation: ABCA1 KO, SR-BI KO, and NPC1 KO cells provide key tools for studying cholesterol efflux, HDL metabolism, and lysosomal cholesterol transport.
· Drug screening and target validation: Adding a test compound (e.g., a PCSK9 inhibitor) to LDLR KO cells allows determination of whether the drug's effect depends on the LDLR pathway.
EDITGENE now offers an off‑the‑shelf/custom knockout cell library covering key genes such as LDLR, CD36, TLR4, ABCA1, PPARG, MYD88, SR‑BI, and NPC1, spanning multiple cell backgrounds including HEK293, HCT116, A549, and HeLa—providing reliable tools for cardiometabolic disease research. Explore the full range of lipoprotein particle stimulus-related KO cell libraries.
Q&A
Q1: I want to study the function of LDLR in specific primary hepatocytes, but that cell type is not listed in your catalog.
We offer custom services. You can provide the target cell line of interest (e.g., human primary hepatocytes or mouse primary hepatocytes), and we will handle the entire process from gene editing to monoclonal validation. In the meantime, you can use our HepG2-derived LDLR KO cells for preliminary experiments.
Q2: I'm planning a high-throughput drug screening and need a large quantity of cells from the same batch. Can you accommodate this?
Yes. All our off-the-shelf cells are supported by a master cell bank and working cell bank system, allowing us to supply large quantities of batch-consistent cells to ensure uniformity and comparability of screening data. We also support customized packaging for 96- or 384-well plates.
Q3: How can I validate the phenotype of KO cells in lipoprotein metabolism?
We recommend the following functional assays:
Dil-LDL uptake assay to assess LDLR function
Oil Red O staining to evaluate foam cell formation
Cholesterol efflux assay to measure ABCA1/ABCG1 activity
Inflammatory cytokine ELISA to assess TLR4/MyD88 pathway response
Western blot to detect LDLR protein expression levels.
Dil-LDL uptake assay to assess LDLR function
Oil Red O staining to evaluate foam cell formation
Cholesterol efflux assay to measure ABCA1/ABCG1 activity
Inflammatory cytokine ELISA to assess TLR4/MyD88 pathway response
Western blot to detect LDLR protein expression levels.
References
[1]. Borén, J., Packard, C. J., & Binder, C. J. (2025). Apolipoprotein B-containing lipoproteins in atherogenesis. Nature Reviews Cardiology.
[2]. Serrano-Pozo, A., Das, S., & Hyman, B. T. (2021). APOE and Alzheimer's disease: advances in genetics, pathophysiology, and therapeutic approaches. The Lancet Neurology, 20(1), 68–80.
[3]. Frambach, S. J. C. M., de Haas, R., Smeitink, J. A. M., Rongen, G. A., Russel, F. G. M., & Schirris, T. J. J. (2020). Brothers in arms: ABCA1- and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacological Reviews, 72(1), 152–190.
[4]. Brandts, J., & Ray, K. K. (2023). Novel and future lipid-modulating therapies for the prevention of cardiovascular disease. Nature Reviews Cardiology, 20(9), 600–616.
Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com
Tag
Share









