















NRAS Mutations in Cancer: Gene Function, Oncogenic Signaling, and Clinically Relevant Variants

NRAS (neuroblastoma RAS viral oncogene homolog) is one of the three members of the RAS proto-oncogene family, together with HRAS and KRAS. It encodes a small GTPase protein with a molecular weight of approximately 21 kDa, which plays a central role in transmitting signaling pathways involved in cell proliferation, differentiation, and survival.
RAS family genes represent some of the most frequently mutated oncogenes in human cancers. Comprehensive analyses estimate that approximately 19% of cancer patients harbor mutations in at least one RAS gene, with KRAS mutations being the most prevalent, followed by NRAS and HRAS mutations.
Compared with KRAS, NRAS mutations exhibit a more restricted distribution among specific tumor types, with particularly high frequencies observed in melanoma, multiple myeloma, and acute myeloid leukemia (AML).
NRAS mutations typically result in constitutive activation of the N-Ras protein, leading to persistent activation of key downstream signaling pathways, including the MAPK and PI3K pathways. As a result, NRAS has long been considered a challenging oncogenic driver to target therapeutically.
To advance functional studies of NRAS mutations and facilitate the development of targeted therapies, NRAS mutant cell lines—particularly isogenic point mutation models generated using CRISPR-based gene editing technologies—have become indispensable research tools.
01
NRAS Gene and Protein Function
The NRAS gene is located on the short arm of human chromosome 1 (1p13.2) and contains seven exons, encoding a protein consisting of 189 amino acids.
Similar to other RAS family members, the N-Ras protein functions as a molecular switch through reversible binding of guanine nucleotides (GTP/GDP).
When bound to GTP, N-Ras adopts an active conformation and interacts with downstream effector proteins, including RAF and PI3K, transmitting signals that promote cell proliferation and survival. Following GTP hydrolysis to GDP, N-Ras returns to an inactive state.
This molecular switch is precisely regulated by two major classes of regulatory proteins:
· Guanine nucleotide exchange factors (GEFs), such as SOS1: Catalyze GDP-to-GTP exchange, thereby activating RAS proteins.
· GTPase-activating proteins (GAPs), such as NF1: Stimulate intrinsic GTP hydrolysis activity of RAS, promoting conversion to the inactive GDP-bound state.
Although the three RAS family members share high structural similarity within their conserved domains, they exhibit significant differences within the hypervariable region (HVR). This region determines their distinct subcellular localization preferences and influences downstream effector interactions.
Compared with KRAS and HRAS, N-Ras exhibits prolonged localization on Golgi and endosomal membranes, which is associated with its unique lipid modification pattern.
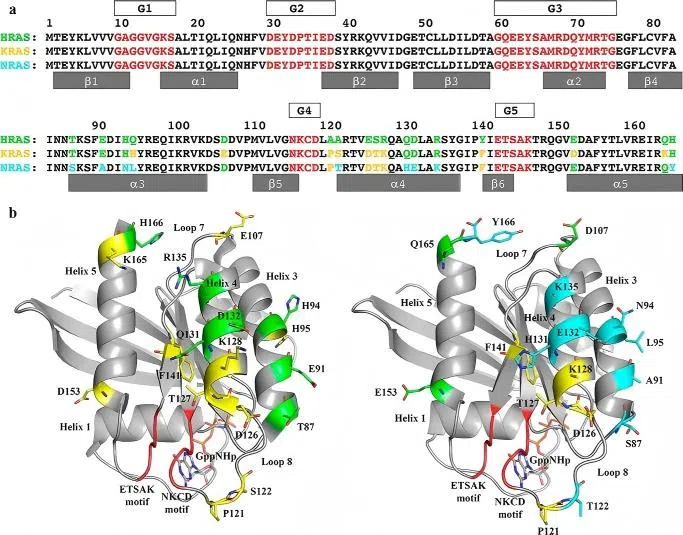 Figure 1. Sequence alignment and structural differences between NRAS and other Ras isoforms
Figure 1. Sequence alignment and structural differences between NRAS and other Ras isoforms 02
NRAS Mutation Hotspots and Cancer Distribution
NRAS mutations are primarily concentrated at three hotspot codons: codon 12 (G12), codon 13 (G13), and codon 61 (Q61).
Among all NRAS mutations, alterations at codon 61 account for approximately 61%–80% of cases, showing a distinct mutation spectrum compared with KRAS and HRAS (Randic et al., 2021).
Different NRAS mutation variants exhibit strong cancer-type preferences. These distribution patterns suggest that individual NRAS variants may possess distinct transforming capabilities within specific cellular contexts, which further influences clinical diagnostic strategies and therapeutic approaches.
2.1 Comparison of NRAS Mutation Variants and Associated Cancer Types
| Variant | Nucleotide Change | Major Associated Cancer Types | Mutation Frequency (among NRAS mutations in specific cancer types) |
|---|---|---|---|
| NRAS Q61R | c.182A>G (CAA→CGA) | Melanoma, thyroid cancer | Accounts for approximately 35–40% of NRAS mutations in melanoma |
| NRAS Q61K | c.181C>A (CAA→AAA) | Melanoma | Accounts for approximately 30–35% of NRAS mutations in melanoma |
| NRAS Q61L | c.182A>T (CAA→CTA) | Melanoma, gastrointestinal cancers | Approximately 10–15% of NRAS mutations |
| NRAS Q61H | c.183A>T/C (CAA→CAT/CAC) | Multiple myeloma, melanoma | A relatively common Q61 variant in myeloma |
| NRAS G12D | c.35G>A | Colorectal cancer, AML | One of the more common NRAS variants in colorectal cancer |
| NRAS G12C | c.34G>T | Various cancer types | Overall frequency is relatively low |
| NRAS G13D | c.38G>A | Colorectal cancer, AML | Less frequent than G12D |
2.2 Melanoma — Q61 Mutations Are Predominant
NRAS-mutant melanoma represents the most common NRAS-driven solid tumor. Approximately 15%–25% of cutaneous melanoma patients harbor activating NRAS mutations.
In melanoma, NRAS mutations are overwhelmingly dominated by codon 61 alterations, which account for approximately 80%–85% of NRAS mutations, whereas G12 and G13 mutations are rare.
Among these variants, NRAS Q61R is the most prevalent single mutation, accounting for approximately 40% of NRAS-mutant melanoma cases, followed by Q61K and Q61L mutations.
NRAS-mutant melanoma and BRAF V600-mutant melanoma are generally mutually exclusive molecular subtypes. NRAS-mutant tumors often exhibit enhanced proliferative activity and invasive characteristics.
Clinically, melanoma harboring NRAS Q61 mutations shows intrinsic resistance to BRAF inhibitors, and available targeted therapeutic options remain limited. As a result, patients with NRAS-mutant melanoma generally have poorer outcomes compared with those carrying BRAF mutations or tumors lacking both BRAF and NRAS alterations.
2.3 Colorectal Cancer — G12/G13 Mutations and Resistance to Anti-EGFR Therapy
NRAS mutations in colorectal cancer are mainly observed at G12, G13, and Q61 hotspots, with an overall incidence of approximately 3%–5%.
Although less frequent than KRAS mutations, NRAS alterations have important clinical implications. Comprehensive testing of RAS status (including both KRAS and NRAS) has become a standard evaluation before first-line therapy for metastatic colorectal cancer.
Regardless of whether the mutation occurs in KRAS or NRAS, the presence of any activating RAS mutation predicts primary resistance to anti-EGFR monoclonal antibodies, including Cetuximab and Panitumumab. Among NRAS variants, NRAS G12D and NRAS G13D are particularly relevant in colorectal cancer.
Both mutations result in persistent activation of the RAS-MAPK signaling pathway downstream of EGFR, rendering EGFR inhibition largely ineffective in blocking downstream signal transmission and directly influencing therapeutic decision-making.
2.4 Multiple Myeloma — Q61 Mutations Account for Approximately 73% of NRAS Alterations
NRAS is one of the most frequently mutated RAS family members in multiple myeloma.
NRAS mutations occur in approximately 20%–25% of newly diagnosed patients and are observed at even higher frequencies in relapsed or refractory disease.
Among NRAS-mutant myeloma cases, Q61 mutations account for approximately 73%.
Overall alterations in the RAS-MAPK pathway, including mutations in KRAS, NRAS, and BRAF, occur in approximately 45%–65% of multiple myeloma cases.
Furthermore, these mutations often display intrapatient clonal heterogeneity, where different tumor cell populations within the same patient carry distinct genetic alterations. This complexity represents a major challenge for targeted therapeutic development.
2.5 Thyroid Cancer — NRAS Mutations Are Associated with Tumor Differentiation Status
NRAS mutations occur in approximately 10%–20% of thyroid tumors, particularly in:
· Follicular thyroid carcinoma (FTC)
· A subset of papillary thyroid carcinoma (PTC)
Compared with tumors carrying the BRAF V600E mutation, thyroid cancers harboring RAS mutations (including NRAS alterations) generally maintain a higher degree of differentiation.
However, these tumors still possess risks of:
· vascular invasion;
· distant metastasis;
· disease progression.
2.6 Acute Myeloid Leukemia (AML) and Other Hematological Malignancies
Approximately 10%–15% of AML patients and a subset of patients with chronic myelomonocytic leukemia (CMML) harbor NRAS mutations.
These mutations mainly occur at:
· G12;
· G13;
· Q61 hotspots.
NRAS mutations frequently coexist with other driver alterations and function as cooperating mutations that contribute to malignant transformation and disease progression.
Among NRAS-mutant cancer models, NRAS G12C represents a relatively rare but clinically important variant.
Although NRAS G12C mutations have been detected sporadically across different cancer types, unlike KRAS G12C, there are currently no approved specific inhibitors targeting NRAS G12C mutations.
This highlights the urgent need for establishing NRAS G12C mutant cell models for mechanistic studies and drug discovery applications.
03
NRAS Oncogenic Signaling Pathways
NRAS mutant proteins promote tumor development primarily through persistent activation of downstream signaling cascades. The major effector pathways regulated by mutant NRAS include the following three signaling axes:
3.1 MAPK Pathway (RAF–MEK–ERK Axis)
Activated N-Ras preferentially interacts with the RAS-binding domain (RBD) of RAF kinases, triggering activation of RAF family members, including CRAF/RAF1.
Activated RAF subsequently phosphorylates and activates MEK1/2, leading to sustained phosphorylation of ERK1/2.
Phosphorylated ERK (pERK) translocates into the nucleus, where it activates multiple transcription factors and induces expression of genes involved in:
· cell cycle progression, such as Cyclin D1;
· cell survival;
· proliferation-associated signaling.
As a result, persistent MAPK pathway activation drives uncontrolled tumor cell proliferation.
Notably, Q61 mutations generally exhibit stronger MAPK pathway activation compared with G12/G13 mutations, primarily because Q61 alterations disrupt both GEF-mediated regulation and GAP-dependent GTP hydrolysis, resulting in prolonged RAS activation.
A study published in Nature in 2025 further revealed that NRAS Q61-mutant cancers rely on the scaffold protein SHOC2 to maintain RAF activation.
Genome-wide CRISPR screening identified SHOC2 as a selective dependency gene in NRAS Q61-mutant cell lines, whereas cells harboring NRAS G12D or G12C mutations showed greater dependency on GRB2 and SHP2 signaling components (Hauseman et al., 2025).
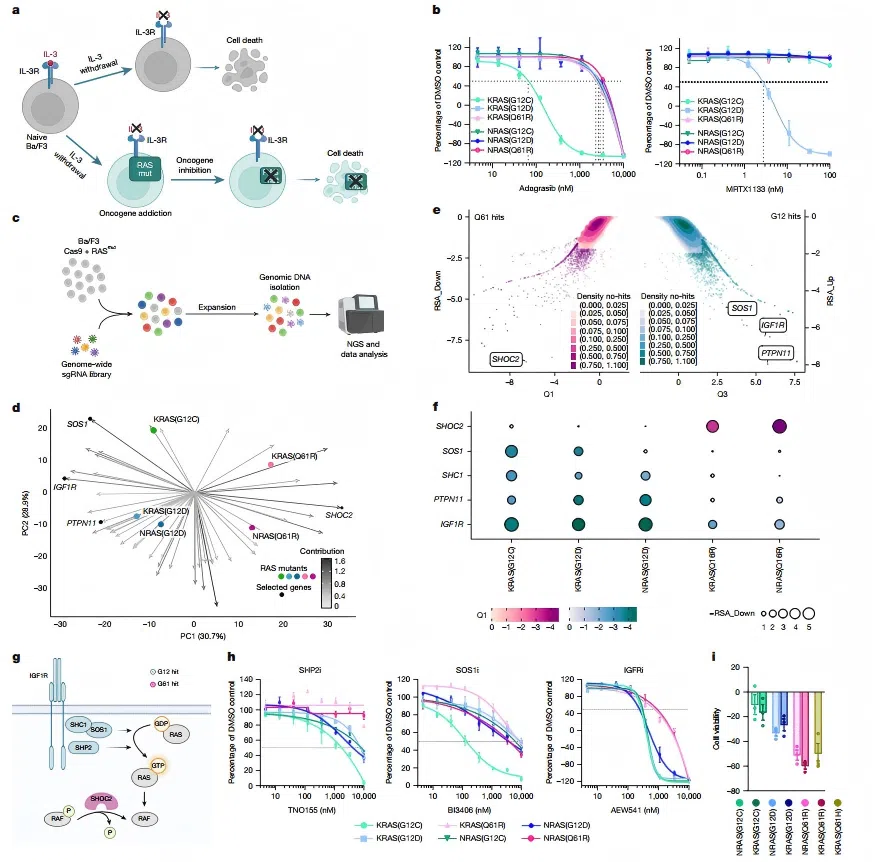 Figure 2. CRISPR knockout screening in Ba/F3 cells identifies mutation-specific RAS dependencies
Figure 2. CRISPR knockout screening in Ba/F3 cells identifies mutation-specific RAS dependencies 3.2 PI3K–AKT–mTOR Pathway
Activated N-Ras can also directly or indirectly stimulate PI3K, primarily through the p110γ/δ catalytic subunits.
PI3K activation promotes the generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3), which subsequently activates:
· AKT kinase;
· downstream mTOR complexes.
The PI3K–AKT–mTOR pathway regulates multiple cellular processes, including:
· glucose uptake;
· protein synthesis;
· cell growth;
· resistance to apoptosis.
Simultaneous activation of both MAPK and PI3K signaling pathways enables NRAS-mutant tumors to maintain survival and proliferation signals even when individual pathway components are pharmacologically inhibited.
This dual pathway activation is considered a major contributor to adaptive resistance against single-target therapies.
3.3 Ral GDS Effector Pathway
In addition to MAPK and PI3K signaling, N-Ras can activate the Ral GDS/RalA-RalB signaling axis.
This pathway contributes to the regulation of:
· vesicle trafficking;
· cytoskeletal remodeling;
· cell migration.
Through these functions, Ral signaling may promote tumor cell invasion and metastatic potential.
04
CRISPR-Engineered NRAS Mutant Cell Models
Naturally occurring cancer cell lines often contain complex background genomic alterations, making it difficult to distinguish the specific biological effects of individual NRAS variants from other coexisting mutations.
In contrast, isogenic NRAS mutant cell models generated using precise genome editing technologies enable direct comparison of different NRAS mutations within an identical genetic background.
These engineered models have become essential tools for investigating NRAS mutation biology and supporting therapeutic development, with key applications including:
· Signaling Mechanism Studies: Quantitative evaluation of how different NRAS variants regulate key signaling nodes, including:
pERK activation;
pAKT activation;
downstream pathway responses.
pAKT activation;
downstream pathway responses.
· Drug Sensitivity Evaluation: Assessment of therapeutic responses under an isogenic genetic background, including experimental compounds targeting:
MEK;
SHOC2;
other components of the RAS signaling network.
SHOC2;
other components of the RAS signaling network.
· Antibody Validation: Use as positive and negative controls to validate the:
specificity;
sensitivity;
mutation recognition capability
sensitivity;
mutation recognition capability
of NRAS mutation-specific antibodies.
· Biomarker Discovery: Identification of mutation-associated molecular signatures through:
transcriptomic analysis;
proteomic profiling;
functional screening approaches.
proteomic profiling;
functional screening approaches.
Based on the Bingo™ prime editing platform (PE7), EDITGENE has established a collection of ready-to-use NRAS mutant cell models covering major hotspot mutations, including: NRAS Q61R, NRAS G12D, NRAS G12C, NRAS G13D.
In addition, EDITGENE a full spectrum of RAS family gene editing solutions, including KRAS and HRAS mutant cell models as well as customized gene engineering services, supporting applications ranging from mechanistic studies to drug screening.
References
[1] Johnson, C. W., Reid, D., et al. (2017). The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. Journal of Biological Chemistry, 292(31), 12981-12993.
[2] Yang, X., & Wu, H. (2024). RAS signaling in carcinogenesis, cancer therapy and resistance mechanisms. Journal of Hematology & Oncology, 17(1), 108.
[3] Dummer, R., Schadendorf, D., et al. (2017). Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncology, 18(4), 435–445.
[4] Hauseman, Z. J., Stauffer, F., Beyer, K. S., et al. (2025). Targeting the SHOC2–RAS interaction in RAS-mutant cancers. Nature.
Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com
Tag
Share









