















G6PD Deficiency and Oxidative Stress: Gene Function, Disease Mechanisms, and Clinically Relevant Variants

What is G6PD? An Overview of the Gene, Enzyme, and Oxidative Stress Pathway
Glucose-6-phosphate dehydrogenase (G6PD) is a critical metabolic enzyme encoded by the G6PD gene. It functions as the first and rate-limiting enzyme in the pentose phosphate pathway (PPP), a metabolic route that serves as a central hub for cellular antioxidant defense and nucleotide synthesis.
G6PD catalyzes the oxidation of glucose-6-phosphate, generating two essential products: NADPH, the primary source of reducing power for cellular redox balance, and ribose-5-phosphate, a key precursor for nucleotide biosynthesis.
NADPH is indispensable for regenerating reduced glutathione (GSH), which directly determines a cell’s ability to neutralize oxidative stress.
Consequently, proper G6PD activity is essential for maintaining erythrocyte integrity and supports the unique metabolic demands of tumor cells. The G6PD gene is located on the X chromosome (Xq28).
More than 230 point mutations have been identified, and approximately 400 million people worldwide carry G6PD variants, making G6PD deficiency one of the most common inherited enzyme disorders (Luzzatto et al., Blood, 2020).
01
G6PD Deficiency: The Genetic Basis of Hemolytic Anemia
What is G6PD Deficiency?
G6PD deficiency is an X-linked genetic disorder caused by functional mutations in the G6PD gene, resulting in partial or complete loss of G6PD enzyme activity. Reduced G6PD activity impairs NADPH production in red blood cells, weakening their defense against oxidative stress.
When exposed to specific triggers—such as primaquine and other drugs, fava beans, or infections—erythrocytes accumulate reactive oxygen species (ROS), leading to oxidative damage and hemolytic anemia.
The genetics of G6PD deficiency are highly polymorphic. The World Health Organization (WHO) classifies G6PD variants into five classes based on residual enzyme activity:
Class I (<10% activity): Associated with chronic nonspherocytic hemolytic anemia
Class II (<10% activity): Associated with acute hemolytic episodes
Class III (10–60% activity): Associated with mild to moderate hemolysis
Class IV/V (normal or increased activity)
Differences among G6PD mutation subtypes in enzyme dimer stability, active site conformation, and substrate affinity determine each variant’s behavior under oxidative stress. This allele-specific nature represents a central challenge in constructing accurate G6PD disease models and assessing drug safety.
The table below summarizes major clinically relevant G6PD variants and their research applications.
| Variant | G6PD mutation site | WHO class | Applications in research |
|---|---|---|---|
| G6PD A− | c.202G>A; c.376A>G (p.Val68Met; p.Asn126Asp) |
Class II | Study of metabolic remodeling and oxidative stress adaptation mechanisms caused by moderate enzyme deficiency |
| G6PD Mediterranean | c.563C>T (p.Ser188Phe) | Class II | Study of the impact of severe enzyme deficiency on the metabolic dependencies of erythrocytes and tumor cells |
| G6PD Canton | c.1376G>T (p.Arg459Leu) | Class II | Hemolytic risk assessment of drugs |
| G6PD Mahidol | c.487G>A (p.Gly163Ser) | Class III | Study of compensatory mechanisms and intervention windows in mild-to-moderate enzyme deficiency |
| G6PD Kaiping | c.1388G>A (p.Arg463His) | Class II | Complementary to Canton for fine dissection of variant-specific functional differences |
| G6PD Seattle | c.844G>C (p.Asp282His) | Class III | As a mild deficiency control for comparing phenotypic thresholds of different deficiency severities |
02
The Dual Role of the G6PD Pathway in Tumor Metabolism
How Do Tumor Cells Hijack the G6PD Pathway?
In contrast to G6PD deficiency, G6PD is significantly overexpressed in many tumors, including hepatocellular carcinoma (HCC), colorectal cancer (CRC), breast cancer, glioma, and pancreatic cancer. Tumor cells upregulate G6PD to increase pentose phosphate pathway (PPP) flux, thereby satisfying two major anabolic needs:
Antioxidant defense: Continuous NADPH production maintains reduced glutathione (GSH), protecting cells against chemotherapy- and radiotherapy-induced oxidative stress.
Rapid proliferation support: Ribose-5-phosphate fuels de novo nucleotide synthesis, while NADPH supports lipid biosynthesis.
p53 Directly Regulates G6PD Enzyme Activity
A key mechanistic link between tumor metabolism and the G6PD gene is p53's direct negative regulation of the enzyme. Wild-type p53 protein binds to G6PD and inhibits its activity, thereby suppressing PPP flux, NADPH generation, and nucleotide synthesis.
In p53-deficient tumor cells, this inhibition is relieved, leading to accelerated PPP activity and a marked proliferative advantage. This mechanism highlights a non-transcriptional pathway through which p53 controls tumor metabolism.
Co-mutation Background Determines G6PD Dependency
Precise interpretation of the G6PD pathway requires consideration of genetic context. G6PD deletion strongly suppresses tumor growth and extends survival in KRAS/LKB1 co-mutant (KL-type) lung cancer, but has minimal effect in KRAS/p53 co-mutant (KP-type) lung cancer.
In KL-type tumors, G6PD loss causes NADPH/GSH imbalance and p53 activation, promoting apoptosis. KP-type tumors, however, are largely insensitive to oxidative stress due to p53 inactivation.
This context-dependent effect underscores the importance of developing genotype-specific isogenic CRISPR cell lines.
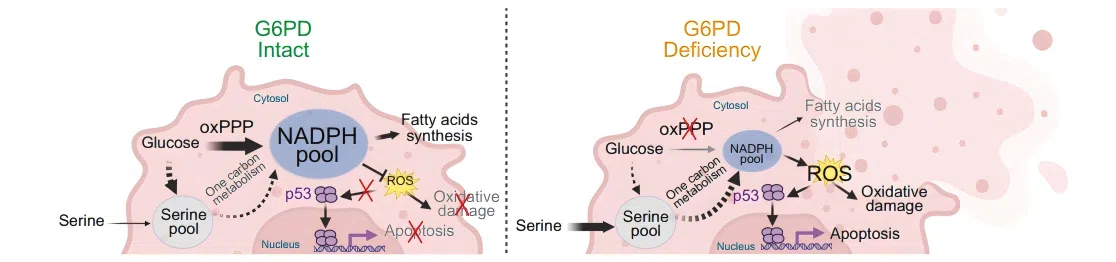 Figure 1. G6PD‑mediated KL lung tumor growth model
Figure 1. G6PD‑mediated KL lung tumor growth model 03
Clinical Application Value of G6PD Disease Models
Colorectal Cancer: G6PD Overexpression and Chemoresistance
Colorectal cancer is one of the most extensively studied solid tumors in G6PD research. Studies in CRC cell lines such as HCT116 have shown that G6PD overexpression protects tumor cells from oxaliplatin-induced oxidative damage by maintaining favorable NADPH/NADP⁺ and GSH/GSSG ratios.
Combined G6PD knockdown and oxaliplatin treatment achieved an 86.7% tumor inhibition rate in xenograft models and was further validated in patient-derived xenograft (PDX) models.
Lung Cancer and Tumor Immunity: G6PD Inhibition Induces Immunogenic Cell Death
The role of the G6PD pathway in tumor immunology is gaining increasing attention. Low G6PD expression correlates with enhanced immune infiltration in the tumor microenvironment. G6PD inhibition can trigger immunogenic cell death (ICD), promoting antigen presentation and immune activation.
In a mouse B16 melanoma model, combining G6PD inhibition with immune checkpoint blockade (ICB) markedly boosted antitumor immunity, providing strong preclinical rationale for G6PD inhibitor–immunotherapy combinations.
Three Major Functional Axes of the G6PD Pathway
In tumors, the G6PD-driven pentose phosphate pathway (PPP) operates through three key functional axes:
NADPH production → Maintains reduced GSH, conferring resistance to chemotherapy- and radiotherapy-induced oxidative stress.
Ribose-5-phosphate supply → Supports de novo purine and pyrimidine synthesis, fueling rapid proliferation.
NADPH-dependent lipid synthesis → Provides fatty acid precursors for cell membrane expansion.
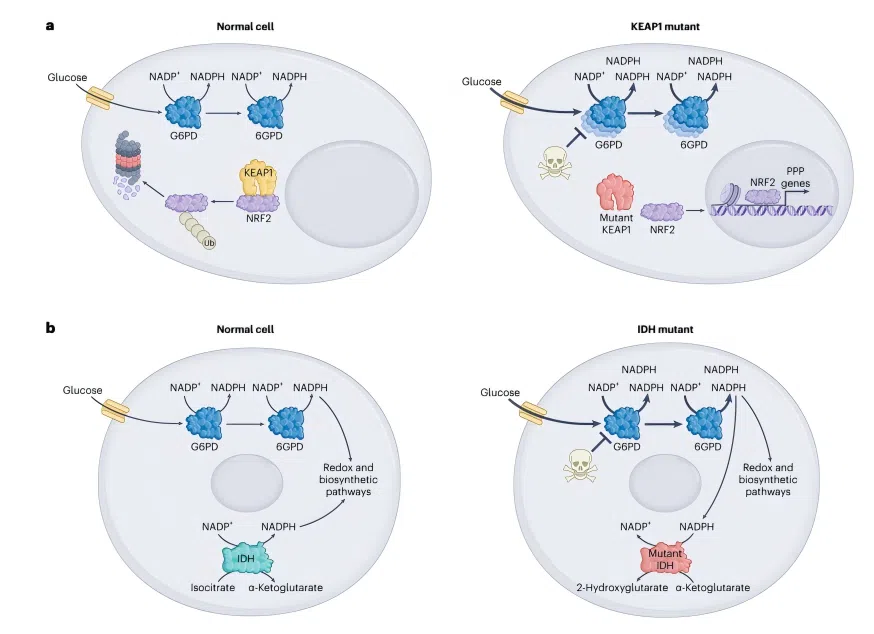 Figure 2. Schematic of the oncogenic landscape of the G6PD pathway in cancer
Figure 2. Schematic of the oncogenic landscape of the G6PD pathway in cancer 04
CRISPR Cell Lines for G6PD Disease Models: Allele-Specific Research Tools
Why Are CRISPR-Edited G6PD Point-Mutant Cell Lines Needed?
Different G6PD variants (e.g., G6PD A⁻, Mediterranean, Canton) exhibit significant allele-specific differences in enzyme activity, NADPH production capacity, and cellular redox phenotypes. Naturally occurring mutant cell lines have complex genetic backgrounds, making it difficult to attribute phenotypes specifically to a single G6PD mutation.
In contrast, CRISPR-mediated precise point-mutation editing enables the creation of isogenic G6PD cell lines in an otherwise identical genetic background. These models allow researchers to:
Dissect mutation function in detail: Parallel comparison of wild-type, heterozygous, and homozygous cells for redox phenotypes.
Study G6PD deficiency genetics: Systematic evaluation of hemolytic risk for individual mutations under exposure to drugs such as rasburicase, primaquine, and bendamustine.
Screen G6PD inhibitors: Assess allele-selective potency of inhibitors (e.g., DHEA, 6-aminonicotinamide [6-AN]).
Investigate drug resistance mechanisms: Model metabolic compensation (e.g., activation of the serine-one-carbon metabolism pathway) in G6PD-deficient tumor cells.
Develop in vivo models: Generate allele-specific mouse xenograft models to evaluate drug efficacy.
05
EDITGENE G6PD Point-Mutation Cell Lines: Ready-to-Use Precision Research Tools
Leveraging the Bingo™ PE7 precision point-editing platform, EDITGENE has successfully generated a panel of CRISPR-edited cell lines harboring various G6PD mutants, covering major clinically relevant variants such as G6PD A− (p.Val68Met, p.Asn126Asp) and G6PD Mediterranean (p.Ser188Phe).
Product Features
Cell background: Standard cell lines including HCT116, K-562, etc.
Validation: Sanger sequencing + monoclonal guarantee
Supply format: In stock, ready to use – no waiting for custom production cycles
Application areas: Genetic research on G6PD deficiency, Functional analysis of oxidative stress, Generation of G6PD disease models, Drug safety assessment
In addition to the G6PD series, EDITGENE also offers off-the-shelf CRISPR Hotspot Precision Mutation Cell Panels, including KRAS, TP53, PIK3CA, EGFR, BRAF, PTEN, ESR1, KIT, IDH1/2, available in multiple cell backgrounds to support the entire research workflow from in vitro screening to in vivo efficacy evaluation.
For a complete list of in-stock items and custom services, please contact EDITGENE customer support.
References
[1]. Luzzatto, L., et al. (2020). Glucose-6-phosphate dehydrogenase deficiency. Blood, 135(17), 1374–1388.
[2]. Jiang, P., et al. (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature Cell Biology, 13(3), 310–316.
[3]. Dong, Y., et al. (2024). Glucose-6-phosphate dehydrogenase maintains redox homeostasis and biosynthesis in LKB1-deficient KRAS-driven lung cancer. Nature Communications, 15(1), 50157.
[4]. Ju, H. Q., et al. (2017). G6PD modulates the sensitivity of colorectal cancer cells to oxaliplatin. Oncogene, 36(43), 5920–5930.
[5]. Zhang, X., et al. (2024). Blockade of glucose-6-phosphate dehydrogenase induces immunogenic cell death and accelerates immunotherapy. Journal for ImmunoTherapy of Cancer, 12(1), e008441.
Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com
Tag
Share









