















[Literature Review] The Misjudgment of Microglia: How TREM2 Mutations Disrupt Brain Balance
点突变细胞
基因编辑

As the primary immune cells of the brain, microglia play a central role in the pathological progression of Alzheimer’s disease (AD). The TREM2 gene is essential for maintaining normal microglial function. Although the T96K mutation has been suggested to enhance certain beneficial aspects of microglial activity, epidemiological studies have paradoxically linked it to an increased risk of Alzheimer’s disease—an inconsistency that has remained unexplained.
Recently, the research team led by Dominika J. Pilat published a study in Neuron titled “The gain-of-function TREM2-T96K mutation increases risk for Alzheimer’s disease by impairing microglial function.” Using CRISPR-Cas9 gene editing, the team successfully generated an AD mouse model carrying the Trem2-T96K mutation. Their work uncovers the biological mechanism by which a so-called “gain-of-function” mutation paradoxically leads to a “loss-of-function” phenotype.

Original link: https://doi.org/10.1016/j.bios.2025.117834
Spotlights
1. Model Construction:
Using CRISPR-Cas9 technology, the researchers generated a Trem2-T96K point mutation knock-in mouse model. They then crossed it with 5xFAD mice to obtain T96K mutant mice with an Alzheimer’s disease background.
2. Mechanistic Insights:
The T96K mutation led to a reduction in the protective soluble TREM2 (sTREM2) and decreased TREM2 expression on the cell surface, impairing the transition of microglia from a homeostatic to a disease-associated state.
3. Implications for TREM2-Targeted Therapy:
The findings suggest that simply enhancing TREM2 activity may not always be beneficial. Factors such as mutation type, sex, and disease stage should be considered to inform precision therapeutic strategies.
The researchers first analyzed genome-wide data from both familial and large case-control cohorts. They confirmed that the T96K mutation is indeed associated with a higher risk of Alzheimer’s disease, with the effect being particularly pronounced in African American (AA) and Hispanic (HISP) populations.
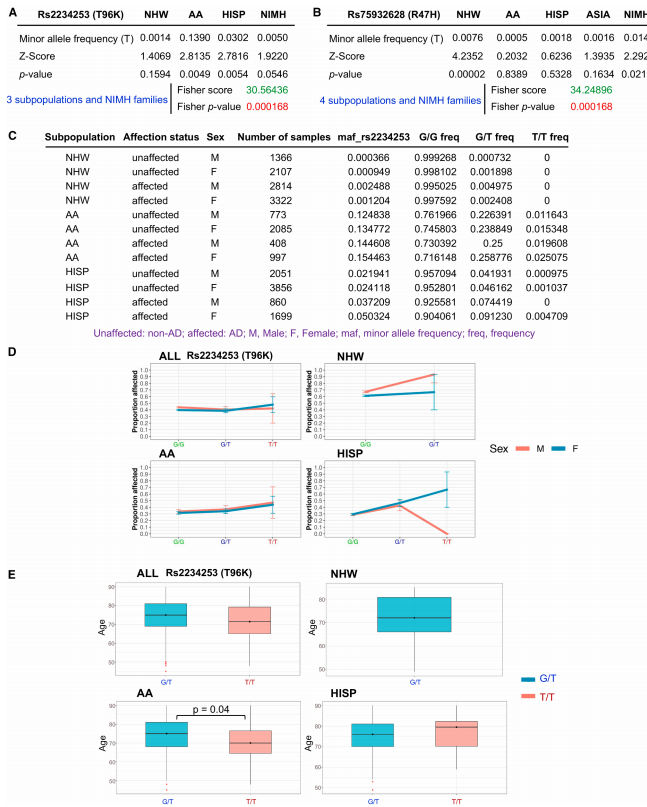
Figure1. Genome-wide data reveal a significant association between the T96K mutation and AD risk
To investigate the impact of the T96K mutation in the diseased brain, the researchers employed CRISPR-Cas9 technology to generate a TREM2-T96K knock-in mouse model that closely mimics the genetic condition found in patients. They further demonstrated that the T96K mutation itself does not alter the overall mRNA expression level of the TREM2 gene.
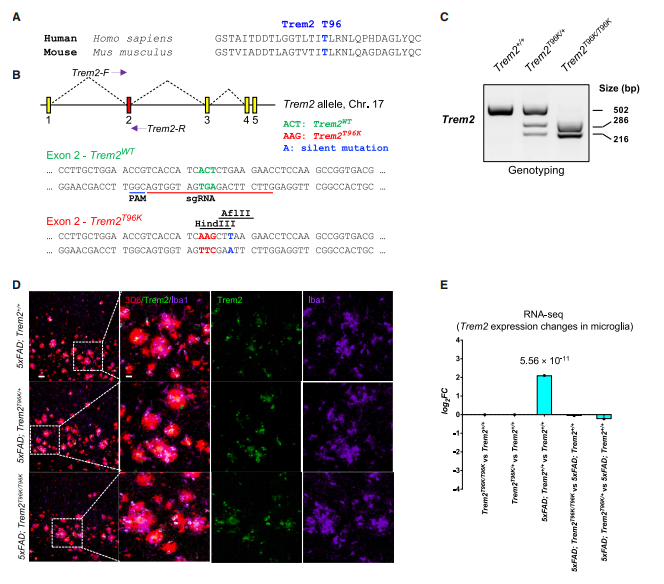
Figure 2. Generation of TREM2-T96K knock-in mice and verification of total TREM2 RNA expression levels
After crossing T96K knock-in mice with the AD model 5xFAD mice, female AD mice carrying either homozygous (T96K/T96K) or heterozygous (T96K/+) mutations showed a significant reduction in the total area of microglial coverage around amyloid plaques in both the cortex and hippocampus. Additionally, the number of microglia clustered around individual plaques was markedly decreased. These findings indicate that the T96K mutation impairs microglial recruitment to Aβ plaques, preventing effective plaque clearance, and this effect is specific to female mice.
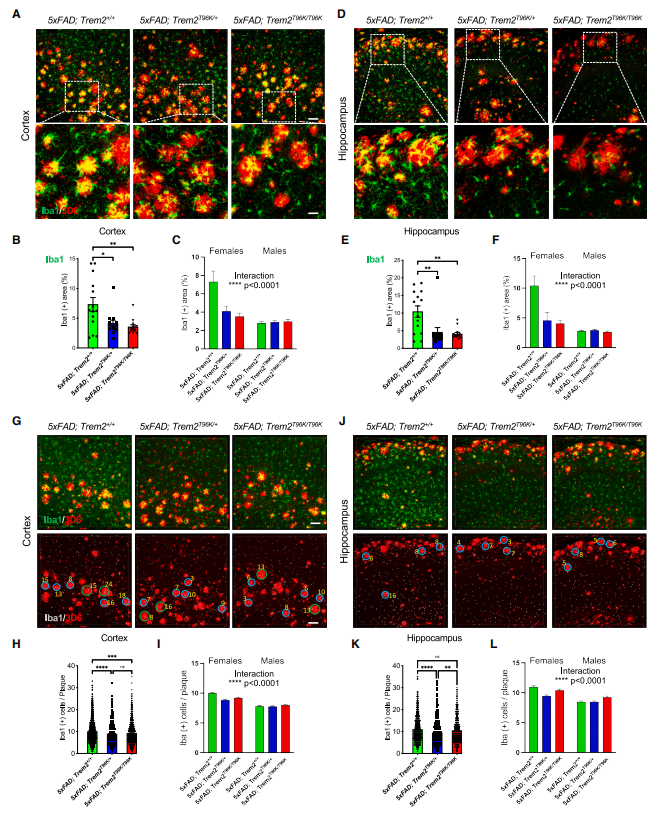
Figure 3. Functional assessment and sex-specific effects of T96K mutant microglia
At the molecular level, analysis of mouse brain tissue proteins and human microglial cultures revealed that the T96K mutation significantly decreases the levels of soluble TREM2 (sTREM2). sTREM2, a fragment released extracellularly after TREM2 cleavage, is critical for intercellular communication and neuroprotection; its reduction indicates that microglia lose this key signaling mediator.
In vitro, T96K/T96K homozygous microglia showed severely impaired uptake of fluorescently labeled Aβ. ELISA quantification of intracellular Aβ levels confirmed this deficiency, demonstrating that the T96K mutation compromises microglial Aβ clearance.
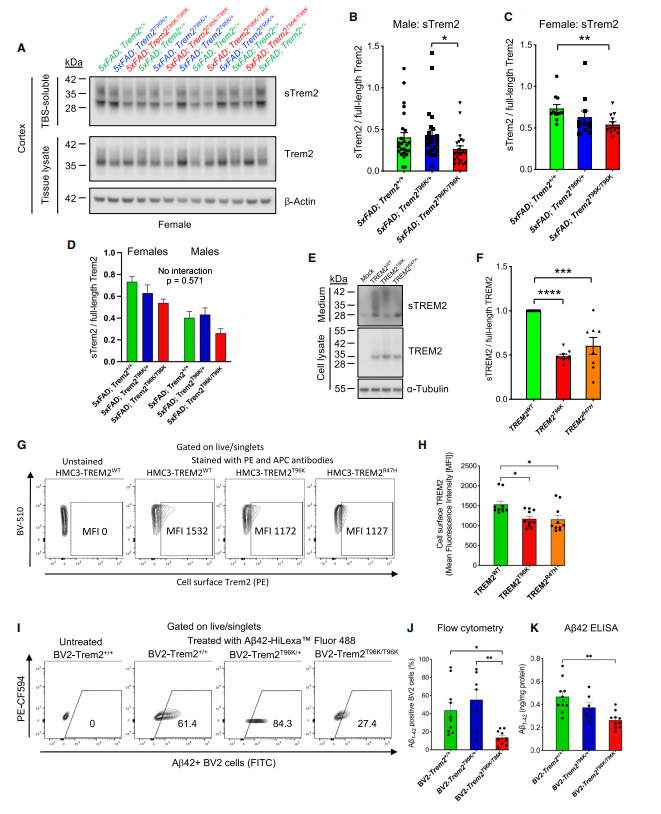
Figure 4. Molecular validation of sTREM2 levels and Aβ clearance function
Single-cell RNA sequencing revealed that the T96K mutation blocks the transition of microglia from a “homeostatic” state to “disease-associated” states.
Using UMAP dimensionality reduction, distinct microglial states were identified: homeostatic (HOM), disease-associated stage 1 (DAM1), and disease-associated stage 2 (DAM2). Notably, in female mutant mice, the proportion of cells transitioning to DAM states was significantly reduced.
Gene expression heatmaps showed that DAM signature genes were downregulated while homeostatic genes remained highly expressed in mutant cells. Trajectory analysis further demonstrated that the “developmental path” toward DAM states was severely disrupted in T96K mutant microglia.
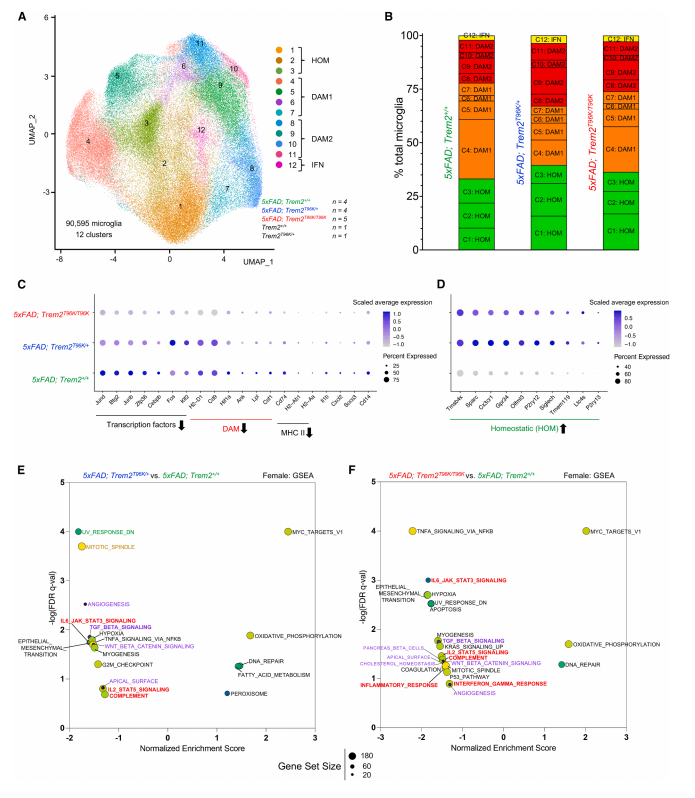
Figure 5. T96K Blocks Microglial Transition to Disease-Associated States
This study provides a new perspective on “gain-of-function” mutations: TREM2-T96K does not simply enhance beneficial activity. Instead, it may overactivate or dysregulate certain signaling pathways, disrupting the precise programs that microglia use to monitor and defend the brain. This dysregulation ultimately manifests as a loss-of-function phenotype, which is particularly pronounced in females.
These findings have profound implications for Alzheimer’s disease drug development. Future therapies targeting TREM2 should not focus solely on activation but must aim for precise modulation to avoid inducing dysfunction similar to that caused by the T96K mutation.
EDITGENE has focused on gene editing for over a decade and has newly developed the Bingo™ platform based on Prime Editing (PE) technology. By combining efficient sgRNA design with a rigorous monoclonal screening system, it provides customized point mutation cell lines of various types for clients.
Recent Blogs
1. [Quality Share]A Comprehensive Guide to CRISPR Detection
3. [Quality Share]RAJI Luciferase Stable Cell Line: Enabling B-Cell Lymphoma Research
Follow us on social media





Contact us
+ 833-226-3234 (USA Toll-free)
+1-224-345-1927 (USA)
info@editxor.com









